
Deep Visual Proteomics and Its Promise for Understanding Diseases

Omics technologies have elevated scientific research to new heights, enabling the high-throughput analysis of thousands of biological molecules from individual and relatively small samples. Technologies such as transcriptomics (RNA), proteomics (proteins), and metabolomics (metabolites) have revolutionized our ability to comprehensively and holistically investigate the landscape of disease states and biological processes at scale. These technologies have been true game-changers, providing unprecedented insights into disease etiology, more effective diagnostics, and informing the development of novel therapeutics.

Perhaps one of the most exciting generational advancements in the omics field has been that of spatial omics. In addition to high-throughput analysis of biological molecules from samples, spatial omics approaches have provided the additional layer of where molecules and the associated changes are happening within cells or tissues.
Given the broad applicability and overall promise of these spatial approaches, it probably comes as little surprise that spatial proteomics was chosen by Nature as their 2024 method of the year. As Nature appropriately states, “Approaches for profiling the spatial proteome in tissues are the basis of atlas-scale projects that are delivering on their promise for understanding biological complexity in health and disease.” While there are many types of “spatial proteomics” methods and techniques, one of the most exciting has been Deep Visual Proteomics (DVP).
DVP was introduced in a 2022 Nature Biotechnology publication from the Mann Lab and their collaborators, and has been actively developed and used since. In the DVP workflow, cell culture or tissue sections are 1) subjected to high-resolution imaging, 2) cells are computationally segmented based on phenotype/morphology, 3) cellular or sub-cellular objects of interest are automatically laser microdissected and collected from a sample section, and 4) they are subjected to mass spectrometry for proteomic analysis. The original DVP workflow overcame the depth limitations of the cells (i.e. single cells didn’t provide enough material for proteomic analysis) by pooling cells with similar phenotypes (morphological features and staining patterns). That limitation was then addressed in a 2023 Nature Methods publication which described the single-cell Deep Visual Proteomics (scDVP) workflow.

A recent and very interesting publication in Nature marks the next major research paper along the Mann lab trajectory of DVP development and utilization. The manuscript is titled “Deep Visual Proteomics maps proteotoxicity in a genetic liver disease” and in it the team outlines the use of their DVP workflow to study the progression of proteotoxicity in patient tissue at the cellular level.
As the name implies, proteotoxicity is cell damage and disruption caused by the accumulation of misfolded and aggregated proteins, and it is a hallmark of diseases including Alzheimer’s disease and Parkinson’s disease. Because of the disease relevance, proteotoxicity is also the focus of therapeutic target development. As the authors point out, DVP is expected to be well suited for studying proteotoxicity in patient samples due to the incorporation of proteomic and visual (stained) data.
In this paper, the authors focused on the genetic disorder, fibrogenic liver disease ⍺1-antitrypsin (AAT) deficiency (AATD). In addition to the clinical relevance, this liver disease is a logical space for the authors to choose given their previous experience with liver tissues, which are highly organized and functionally repetitive tissue that make it well suited for DVP applications (as outlined in their previous DVP 2023 paper). The authors were able to collect a cohort of FFPE biopsies and liver explant tissue samples from patients encompassing the full range of fibrosis severity. Age was similar across the patient cohort.
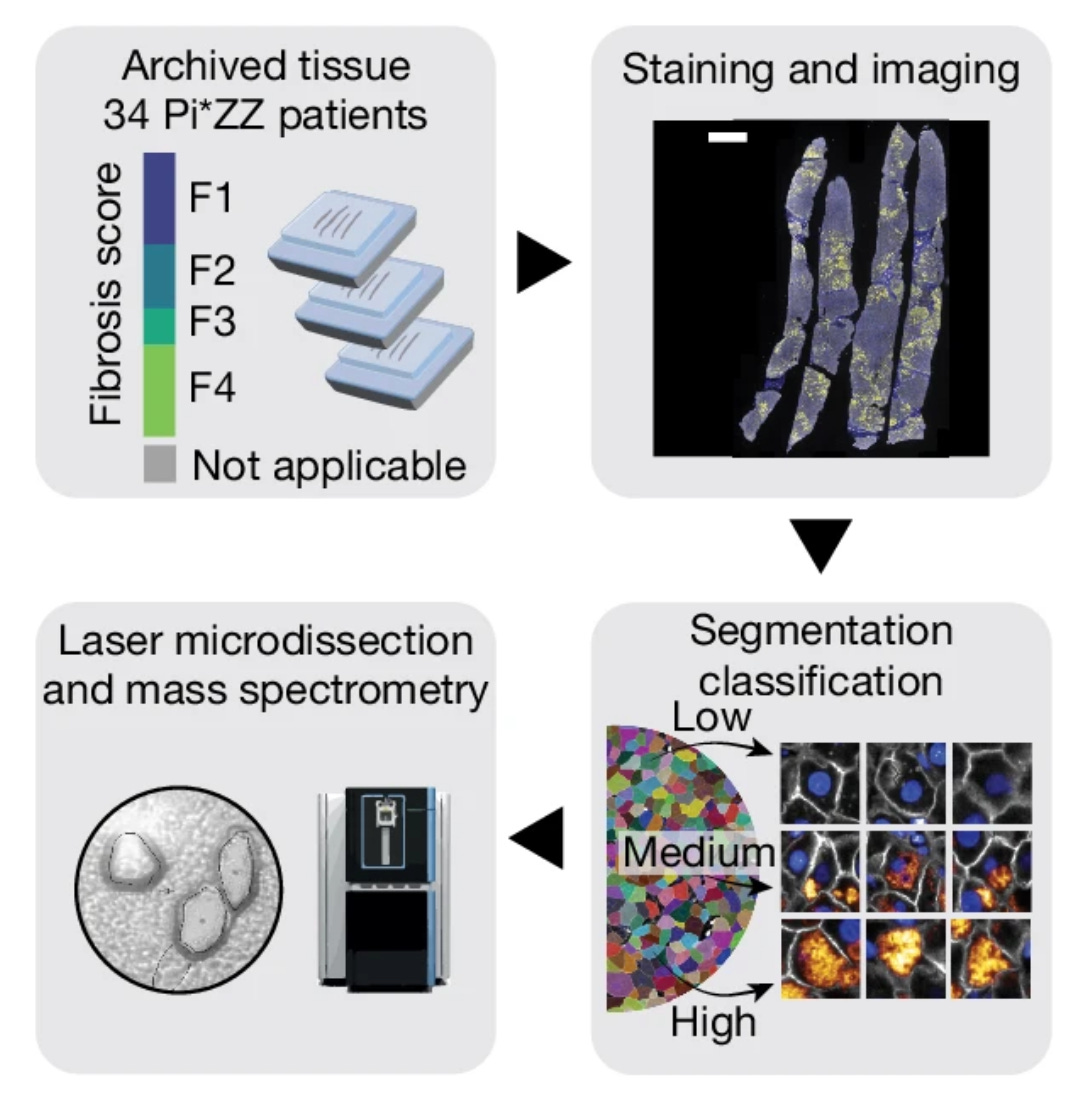
The authors used the samples to create proteomic maps of the tissues across fibrosis severity scores, and the cells were also stratified into low, moderate, and high aggregate load groups based on the AAT intensity. Across the progression of AAT load, the authors observed markedly different proteomes, including elevation of proteins related to unfolded protein response and a reduction in transcription and translation machinery.
From here the authors identified protein levels that correlated with AAT accumulation (proteins that they call “AAT followers”), and they also categorized proteins into groups as “early” and “late” responders to proteotoxic stress caused by (and indicated by) AAT accumulation. This yielded insight into potential cellular mechanisms associated with the progression of disease AAT accumulation. They also demonstrate the mapping of tissue at the single-cell level and by morphology, which demonstrated the broad capabilities and overall adaptability of the technology for demystifying disease cellular mechanisms across disease tissue topography.

Overall this paper provided additional insight into 1) the ⍺1-antitrypsin (AAT) deficiency disease while also 2) demonstrating the utility of the DVP framework for understanding diseases at the spatiotemporal molecular level. From a disease perspective, the authors validated their systems by identifying known disease mechanisms while at the same time identifying new, additional proteins and pathways for further investigation. We can expect this paper to setup additional studies to more deeply understand and validate the newly identified proteins and pathways. We could also expect researchers to adopt this framework to studying their own sets of tissue samples, or even to study other related diseases. The insights could also lead to therapeutic hypotheses and development.
From a methodological perspective, this paper represents a significant step forward for the DVP framework, and will certainly setup more applications in the future. The authors notably present single-cell DVP for FFPE tissues, which up to now has only been reported to be used effectively for frozen tissue sections. The authors note that throughput will be an area of future development, but in its current state this framework holds great promise for studying disease using FFPE tissue sections, which are quite commonly available. There are many interesting analytical and methodological advances outlined in the paper, which can be fully reviewed in the manuscript (which is a short and straightforward read).
Altogether this was a super interesting read, both from the disease biology perspective and the methods+analytical perspective. As we mentioned above, this blog post is just a high-level summary of the paper and any interested readers should dig into the manuscript for more details.
And as always, if we missed anything or if you have any thoughts/insights from the paper, be sure to share in the comments!