
Fighting Pathogens With AI-Predicted Protein Structures
A Deep Dive Into Recently Published Research in PLOS Computational Biology

Microbial pathogens remain a major and constant threat to human health. Antibiotics and antimicrobial drugs have shifted the paradigm of pathogenic disease treatment, but the rise of antimicrobial resistance necessitates continued development of therapeutic approaches. Vaccine development also remains an important pillar to the fight against pathogenic disease, and we continue to require new and innovative approaches as was especially evident during the COVID-19 pandemic.
One important component to antimicrobial drug and vaccine development is understanding the interactions between humans and pathogens at the protein level (i.e. protein-protein interactions). While there are multiple approaches to studying protein-protein interactions, a particularly powerful approach is to determine, visualize, and examine their 3-Dimensional structures. Understanding the structures of proteins, and obtaining highly accurate structural models of their interactions, can be important for therapeutic development and recent advances in artificial intelligence methods such as the AlphaFold models have made such structural analysis increasingly accessible.
A recent article in PLOS Computational Biology titled “AI-first structural identification of pathogenic protein target interfaces” by Mihkel Saluri et al examined the use of contemporary AI methods to predict the structures of human-pathogen protein-protein interactions. This is an understudied space in which only a small fraction of experimentally observed interactions have been structurally modeled. In this publication the authors outlined an AI-driven framework for host-pathogen protein interaction structural modeling, which could have implications for vaccine and drug development.
In this blog post we will continue our trend on protein analytics, and will review the advances outlined in this publication. We will also discuss some of the impactful applications that could follow. As we dig in we will quickly realize that the Saluri et al paper follows a familiar outline in which they 1) setup their analytical framework and parameters, 2) apply their framework to gain novel insights and generate new interesting hypotheses, and then 3) perform some experimental follow-up to validate one of their in silico findings.
1) Setup: Establishing FoldDock
In this paper the authors made heavy use of the FoldDock pipeline, which was developed by the corresponding author Patrick Bryant and colleagues, and outlined in previous publications and code repositories (see references and further reading below). As the name suggests, the FoldDock pipeline accurately models the folding and docking of two proteins simultaneously, leveraging a combination of AlphaFold2 (for modeling protein structures) and multiple sequence alignment. It’s been used across domains of life including eukaryotic and bacteria, but has not yet been applied to host-pathogen protein-protein interactions to this level of robustness.
The authors began their most recent paper by establishing the use of the FoldDock pipeline, as well as comparing it to the use of FoldDock with templates and to the use of a similar tool AlphaFold-Multimer (AFM). The team found that the use of FoldDock with templates resulted in increased false positives, and that FoldDock alone performed more favorably across their benchmarking. Armed with this evidence of superior performance and improved discriminatory ability, the authors proceeded to use only FoldDock for the remainder of the paper.
2) Application: Generating Novel Protein Structures
Once the use of FoldDock had been established, the authors turned their attention to identifying novel host-pathogen protein-protein interactions. To accomplish this, the team leveraged the Host-Pathogen Interaction Database 3.0 and specifically identified the pathogenic microbes with the greatest prevalence of interactions with humans. The resulting pathogens and the number of interactions are highlighted in their figure below, and there was a total of 9,576 interactions that they identified.
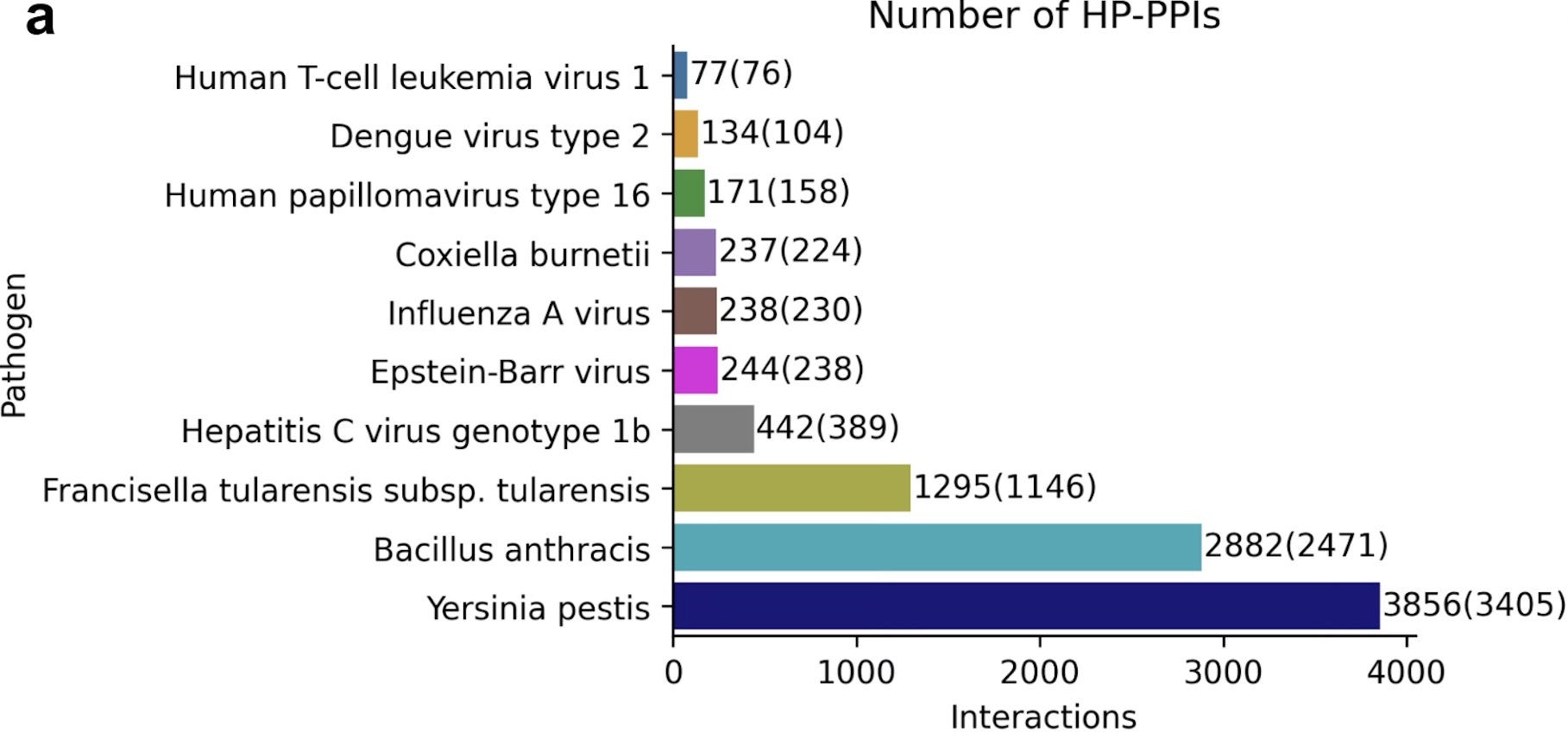
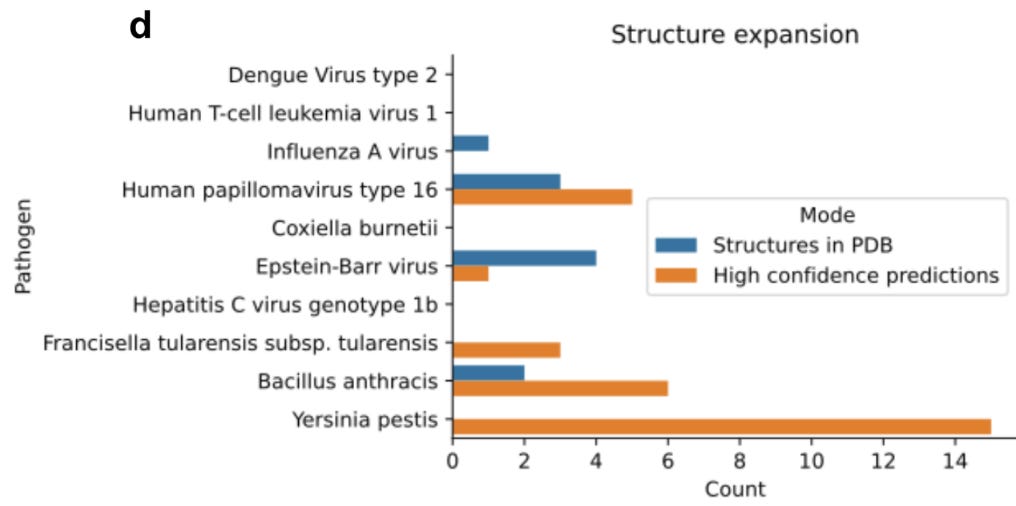
The team used their FoldDock pipeline (with some additional technical tuning) to identify 30 high-quality predictions of protein-protein interaction structures between pathogens and humans (orange in the figure below). The database originally had only 10 known protein-protein complex structures (blue in the figure below), and interestingly none of those were accurately predicted by FoldDock (highlighting an important need that remains for the other “traditional” techniques). This means that the FoldDock application increased the total number of structures four-fold (to a total of 40) and provided some high confidence predictions for some pathogens with no previously known structures (e.g. Yersinia Pestis).
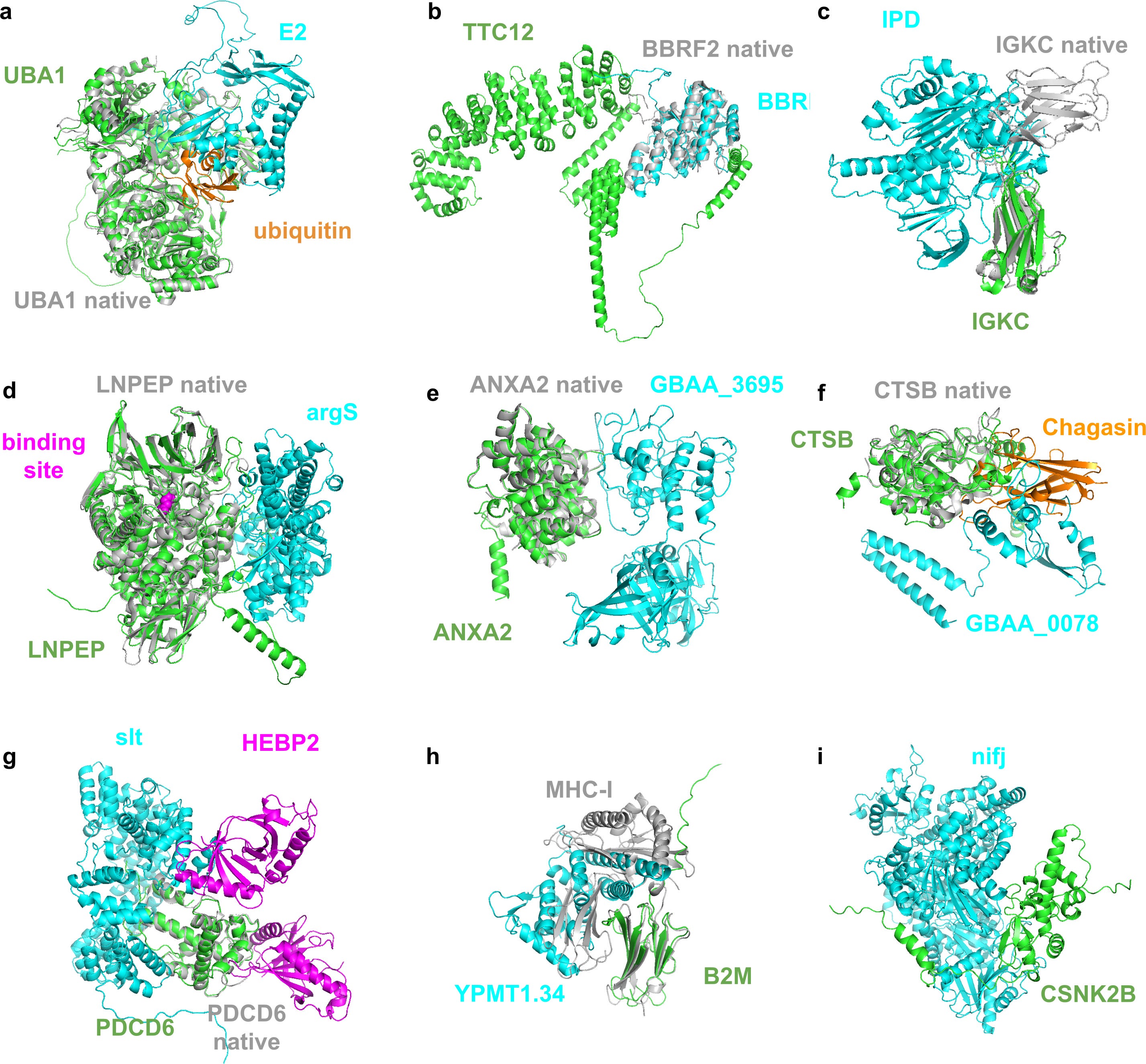
The authors went on to provide a deeper dive into the nine highest quality predictions of the 30 mentioned above (see figure below). Interested readers can go through the manuscript for the team’s more detailed review. We can however highlight here that some of these interactions appear to include proteins of unknown UniProt functions/definitions, which highlights the utility of this FoldDock framework for providing additional insights and hypotheses into poorly defined proteins which may warrant further investigation.
3) Experimental Validation: Using Mass Spectrometry To Confirm Computational Predictions
Finally the authors dug deeper into one of the nine high-quality protein-protein complex predictions; that of F. tularensis IPD and IGKC. The authors mentioned that this potential interaction had already been identified previously through a yeast two-hybrid screen and was therefore a good candidate for further investigation. The authors leveraged native Mass Spectrometry and recombinantly produced proteins from E. coli to investigate the validity of the predicted relationship. The resulting spectra did in fact support the structural predictions of the FoldDock pipeline, thereby providing greater confidence in the outlined framework for accurately predicting the structures of host-pathogen protein-protein interactions.
Implications & Future Directions
Overall this is a rather quick read that is at the same time dense with data and information. Like a lot of good papers however, that value goes beyond the mechanistic scientific insights. While it’s certainly interesting to see newly predicted interaction structures that warrant followup investigation, this framework provides a great launchpad for further, deeper analysis and research. As the authors put it, this paper is providing an “AI-guided framework for host-pathogen structure prediction, aimed at uncovering novel interactions of functional and clinical relevance”.
As the authors point out, the most obvious application of this framework is perhaps within the fields of infectious diseases and vaccine development. Accurate structural prediction can play an important role in understanding the underlying mechanisms of host-pathogen interactions, especially when it involves unknown or understudied proteins. Such structural predictions can also play a critical role in rational drug design as well as vaccine development.
Another valuable application of this technology could be for the triage of potential targets of interest from large, systems biology data analyses or high-throughput assays. Often these types of screens and studies can provide large lists of targets and “hits” for followup, and this structural modeling approach could provide a valuable approach to funneling down such a list, and removing false positives or “hits” with low likelihood of success.
Finally, while the application to human-pathogen interactions is very interesting and obviously very important, it will also be fascinating to see how this approach might be applied to host-pathogen interactions in animals and agriculture (a future direction also noted by the authors). Infectious disease prevention, vaccines, and treatments are of the utmost importance for livestock farming, and it is equally important in agriculture and the successful farming of crops. The importance of these fields to our food supply, the scale involved, and the ability of pathogens to spread quickly and efficiently in these environments makes innovation both challenging and necessary. The framework may also have valuable applications for protein-protein interactions between pathogenic microbes and insects, from an insect control perspective.
Conclusions
This manuscript is an interesting read which outlines an analytical framework with a variety of interesting and potentially valuable avenues of application. In this blog post we reviewed how the authors established their analytical framework, applied their framework to gain novel insights, and then performed some experimental follow up to validate one of their in silico findings. We then reflected on the many ways this could be used in drug and vaccine development, as well as applications beyond human health. Of course we are only reviewing the topic at a high level in this post, and interested readers should certainly read the manuscript for details. Resources for further reading, as well as the subject manuscript of this post, can be found below.
Further Reading & References
Improved prediction of protein-protein interactions using AlphaFold2. 2022.
Towards a structurally resolved human protein interaction network. 2023.
Accurate structure prediction of biomolecular interactions with AlphaFold 3. 2024.
AI-first structural identification of pathogenic protein target interfaces. 2025.